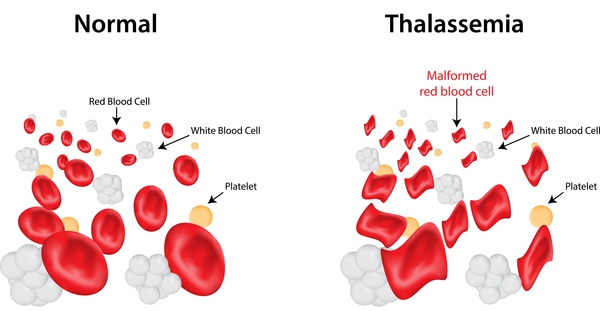
Thalassemia is an inherited blood disorder in which the body is unable to make adequate hemoglobin. Most of our physical characteristics are inherited through the genes we take from our parents; for example, the shape of our nose, the colour of our skin and eyes. We also inherit our hemoglobin (Hb) type from our parents, through the genes. Hemoglobin is present in the red cells and is made from proteins. These proteins consist of alpha(α) and beta(β) chains. Normally 2 alpha and 2 beta chains are essential to form hemoglobin. Imbalances in these chains results in reduced red cell survival. The amount of beta and alpha chains a person makes is controlled by the hemoglobin gene they inherit from their parents Normally red cells survive for 120 days but in Thalassemia red cell survival is reduced. Survival of Thalassemia patients depends upon repeated blood transfusion and costly medicines. In our country beta Thalassemia is very common and the term Thalassemia denotes beta Thalassemia.
Prevention is better than cure, Thalassemia is a completely preventable disorder.It is suggested the following prevention policies can be adopted to reduce affected births by doing blood tests (HbA2 estimation) and save resources so that the best possible treatment can be made available for already existing cases.
There are an estimated 60-80 million people in the world who carry the beta thalassemia trait alone. This is a very rough estimate and the actual number of thalassemia Major patients is unknown due to the prevalence of thalassemia in less developed countries in the Middle East and Asia. Countries such as India, Pakistan and Iran are seeing a large increase of thalassemia patients due to lack of genetic counseling and screening. Most of them die in early life, often without a diagnosis or because of inadequate treatment. There is growing concern that thalassemia may become a very serious problem in the next 50 years, one that will burden the world’s blood centre supplies and the health system in general.
We use cookies to improve your experience on our site. By using our site, you consent to cookies.
Manage your cookie preferences below:
Essential cookies enable basic functions and are necessary for the proper function of the website.
These cookies are needed for adding comments on this website.
Google Analytics is a powerful tool that tracks and analyzes website traffic for informed marketing decisions.
Google reCAPTCHA helps protect websites from spam and abuse by verifying user interactions through challenges.
Marketing cookies are used to follow visitors to websites. The intention is to show ads that are relevant and engaging to the individual user.
Google Maps is a web mapping service providing satellite imagery, real-time navigation, and location-based information.
Service URL: policies.google.com (opens in a new window)